Table of Contents
Relationship of Drug Concentrations to Drug Response
The initiation of drug therapy starts with the manufacturer’s recommended dosage regimen that includes the drug dose and frequency of doses (eg, 100 mg every 8 hours). Due to individual differences in the patient’s genetic makeup or pharmacokinetics, the recommended dosage regimen drug may not provide the desired therapeutic outcome. The measurement of plasma drug concentrations can confirm whether the drug dose was subtherapeutic due to the patient’s individual pharmacokinetic profile (observed by low plasma drug concentrations) or was not responsive to drug therapy due to genetic differences in receptor response.
response.
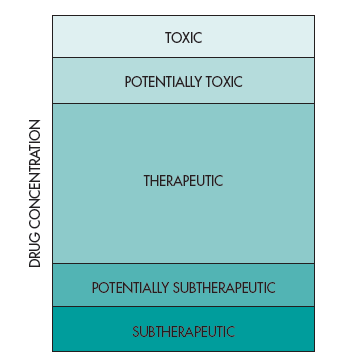
In this case, the drug concentrations are in the therapeutic range but the patient does not respond to drug treatment. Figure 1-2 shows that the concentration of drugs in the body can range from subtherapeutic to toxic. In contrast, some patients respond to drug treatment at lower drug doses that result in lower drug concentrations. Other patients may need higher drug concentrations to obtain a therapeutic effect, which requires higher drug doses. It is desirable that adverse drug responses occur at drug concentrations higher relative to the therapeutic drug concentrations, but for many potent drugs, adverse effects can also occur close to the same drug concentrations as needed for the therapeutic effect.
Pharmacodynamics
Pharmacodynamics refers to the relationship between the drug concentration at the site of action (receptor) and pharmacologic response, including biochemical and physiologic effects that influence the interaction of the drug with the receptor. The interaction of a drug molecule with a receptor causes the initiation of a sequence of molecular events resulting in a pharmacologic or toxic response. Pharmacokinetic-pharmacodynamic models are constructed to relate plasma drug level to drug concentration at the site of action and establish the intensity and time course of the drug.
Drug exposure and drug response
Drug exposure refers to the dose (drug input to the body) and various measures of acute or integrated drug concentrations in plasma and other biological fluid (eg, Cmax, Cmin, Css, AUC) (FDA Guidance for Industry, 2003). Drug response refers to a direct measure of the pharmacologic effect of the drug. Response includes a broad range of endpoints orbiomarkers ranging from the clinically remote biomarkers (eg, receptor occupancy) to a presumed mechanistic effect (eg, ACE inhibition), to a potential or accepted surrogate (eg, effects on blood pressure, lipids, or cardiac output), and to the full range of short-term or long-term clinical effects related to either efficacy or safety. Toxicologic and efficacy studies provide information on the safety and effectiveness of the drug during development and in special patient populations such as subjects with renal and hepatic insufficiencies. For many drugs, clinical use is based on weighing the risks of favorable and unfavorable outcomes at a particular dose. For some potent drugs, the doses and dosing rate may need to be titrated in order to obtain the desired effect and be tolerated.
Toxicokinetics and clinical toxicology
Toxicokinetics is the application of pharmacokinetic principles to the design, conduct, and interpretation of drug safety evaluation studies (Leal et al, 1993) and in validating dose-related exposure in animals. Toxicokinetic data aid in the interpretation of toxicologic findings in animals and extrapolation of the resulting data to humans. Toxicokinetic studies are performed in animals during preclinical drug development and may continue after the drug has been tested in clinical trials.
Clinical toxicology is the study of adverse effects of drugs and toxic substances (poisons) in the body. The pharmacokinetics of a drug in an overmedicated (intoxicated) patient may be very different from the pharmacokinetics of the same drug given in lower therapeutic doses. At very high doses, the drug concentration in the body may saturate enzymes involved in the absorption, biotransformation, or active renal secretion mechanisms, thereby changing the pharmacokinetics from linear to nonlinear pharmacokinetics. Drugs frequently involved in toxicity cases include acetaminophen, salicylates, opiates (eg, morphine), and the tricylic antidepressants (TCAs). Many of these drugs can be assayed conveniently by fluorescence immunoassay (FIA) kits.