Table of Contents
Measurement of drug concentrations
Because drug concentrations are an important element in determining individual or population pharmacokinetics, drug concentrations are measured in biological samples, such as milk, saliva, plasma, and urine. Sensitive, accurate, and precise analytical methods are available for the direct measurement of drugs in biologic matrices. Such measurements are generally validated so that accurate information is generated for pharmacokinetic and clinical monitoring. In general, chromatographic and mass spectrometric methods are most frequently employed for drug concentration measurement, because chromatography separates the drug from other related materials that may cause assay interference and mass spectrometry allows the detection of molecules or molecule fragments based on their mass-to-charge ratio.
Sampling of Biologic Specimens
Only a few biological specimens may be obtained safely from the patient to gain information as to the drug concentration in the body. Invasive methods include sampling blood, spinal fluid, synovial fluid, tissue biopsy, or any biologic material that requires parenteral or surgical intervention in the patient. In contrast, noninvasive methods include a sampling of urine, saliva, feces, expired air, or any biological material that can be obtained without parenteral or surgical intervention.
The measurement of drug and metabolite concentration in each of these biologic materials yields important information, such as the amount of drug retained in, or transported into, that region of the tissue or fluid, the likely pharmacologic or toxicologic outcome of drug dosing, and drug metabolite formation or transport. Analytical methods should be able to distinguish between protein-bound and unbound parent drugs and each metabolite, and the pharmacologically active species should be identified. Such distinctions between metabolites in each tissue and fluid are especially important for the initial pharmacokinetic modeling of a drug.
Drug Concentrations in Blood, Plasma, or Serum
Measurement of drug and metabolite concentrations (levels) in the blood, serum, or plasma is the most direct approach to assessing the pharmacokinetics of the drug in the body. Whole blood contains cellular elements including red blood cells, white blood cells, platelets, and various other proteins, such as albumin and globulins. In general, serum or plasma is most commonly used for drug measurement. To obtain serum, whole blood is allowed to clot and the serum is collected from the supernatant after centrifugation. Plasma is obtained from the supernatant of centrifuged whole blood to which an anticoagulant, such as heparin, has been added. Therefore, the protein content of serum and plasma is not the same. Plasma perfuses all the tissues of the body, including the cellular elements in the blood. Assuming that a drug in the plasma is in dynamic equilibrium with the tissues, then changes in the drug concentration in plasma will reflect changes in tissue drug concentrations. Drugs in the plasma are often bound to plasma proteins, and often plasma proteins are filtered from the plasma before drug concentrations are measured. This is the unbound drug concentration. Alternatively, drug concentration may be measured from unfiltered plasma; this is the total plasma drug concentration. When interpreting plasma concentrations, it is important to understand what type of plasma concentration the data reflect.
Plasma Drug Concentration–Time Curve
The plasma drug concentration (level)–time curve is generated by obtaining the drug concentration in plasma samples taken at various time intervals after a drug product is administered. The concentration of drug in each plasma sample is plotted on rectangularcoordinate graph paper against the corresponding time at which the plasma sample was removed. As the drug reaches the general (systemic) circulation, plasma drug concentrations will rise up to a maximum if the drug was given by an extravascular route. Usually, absorption of a drug is more rapid than elimination. As the drug is being absorbed into the systemic circulation, the drug is distributed to all the tissues in the body and is also simultaneously being eliminated. Elimination of a drug can proceed by excretion, biotransformation, or a combination of both. Other elimination mechanisms may also be involved, such as elimination in the feces, sweat, or exhaled air.
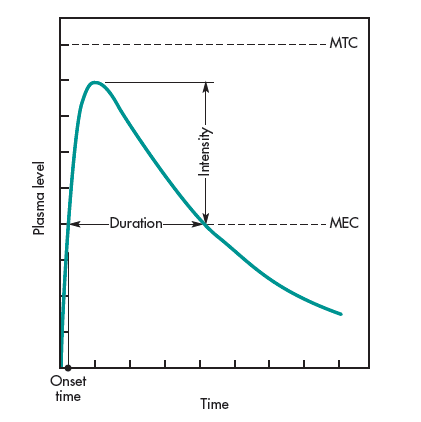
oral administration of a drug.
The relationship of the drug level–time curve and various pharmacologic parameters for the drug are shown in Fig. 1-3. MEC and MTC represent the minimum effective concentration and minimum toxic concentration of drugs, respectively. For some drugs, such as those acting on the autonomic nervous system, it is useful to know the concentration of drug that will just barely produce a pharmacologic effect (ie, MEC). Assuming the drug concentration in the plasma is in equilibrium with the tissues, the MEC reflects the minimum concentration of drug needed at the receptors to produce the desired pharmacologic effect. Similarly, the MTC represents the drug the concentration needed to just barely produce a toxic effect. The onset time corresponds to the time required for the drug to reach the MEC. The intensity of the pharmacologic effect is proportional to the number of drug receptors occupied, which is reflected in the observation that higher plasma drug concentrations produce a greater pharmacologic response, up to a maximum. The duration of drug action is the difference between the onset time and the time for the drug to decline back to the MEC.
The therapeutic window is the concentrations between the MEC and the MTC. Drugs with a wide therapeutic window are generally considered safer than drugs with a narrow therapeutic window. Sometimes the term therapeutic index is used. This term refers to the ratio between toxic and therapeutic doses.
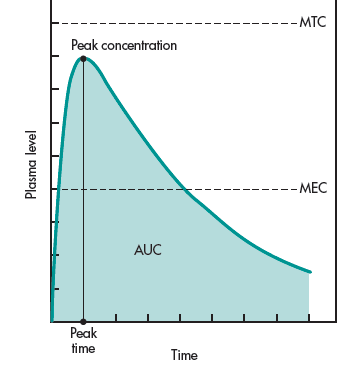
and concentration. The shaded portion represents the AUC
(area under the curve).
In contrast, pharmacokinetics can also describe the plasma level–time curve in terms of such pharmacokinetic terms as peak plasma level (Cmax), time for peak plasma level (Tmax), and area under the curve, or AUC (Fig. 1-4). The time for peak plasma level is the time of maximum drug concentration in the plasma and is a rough marker of the average rate of drug absorption. The peak plasma level or maximum drug concentration is related to the dose, the rate constant for absorption, and the elimination constant of the drug. The AUC is related to the amount of drug absorbed systemically.
Drug Concentrations in Tissues
Tissue biopsies are occasionally removed for diagnostic purposes, such as the verification of a malignancy. Usually, only a small sample of tissue is removed, making drug concentration measurement difficult. Drug concentrations in tissue biopsies may not reflect drug concentration in other tissues nor the drug concentration in all parts of the tissue from which the biopsy material was removed. For example, if the tissue biopsy was for the diagnosis of a tumor within the tissue, the blood flow to the tumor cells may not be the same as the blood flow to other cells in this tissue. In fact, for many tissues, blood flow to one part of the tissues need not be the same as the blood flow to another part of the same tissue. The measurement of the drug concentration in tissue biopsy material may be used to ascertain if the drug reached the tissues and reached the proper concentration within the tissue.
Drug Concentrations in Urine and Feces
Measurement of drugs in urine is an indirect method to ascertain the bioavailability of a drug. The rate and extent of drug excreted in the urine reflects the rate and extent of systemic drug absorption.
Measurement of the drug in feces may reflect drug that has not been absorbed after an oral dose or may reflect drug that has been expelled by biliary secretion after systemic absorption. Fecal drug excretion is often performed in mass balance studies, in which the investigator attempts to account for the entire dose given to the patient. For a mass balance study, both urine and feces are collected and their drug content measured. For certain solid oral dosageforms that do not dissolve in the gastrointestinal tract but slowly leach out the drug, the fecal collection is performed to recover the dosage form. The undissolved dosage form is then assayed for the residual drugs.
Drug Concentrations in Saliva
Saliva drug concentrations have been reviewed for many drugs for therapeutic drug monitoring (Pippenger and Massoud, 1984). Because only freedrug diffuses into the saliva, saliva drug levels tend to approximate free drug rather than total plasma drug concentration. The saliva/plasma drug concentration ratio is less than 1 for many drugs. The saliva/ The plasma drug concentration ratio is mostly influenced by the pKa of the drug and the pH of the saliva. Weak acid drugs and weak base drugs with pKa significantly different than pH 7.4 (plasma pH) generally have a better correlation to plasma drug levels. The saliva drug concentrations were taken after equilibrium with the plasma drug concentration generally provide a more stable indication of drug levels in thebody. The use of salivary drug concentrations as a the therapeutic indicator should be used with caution and preferably as a secondary indicator.
Forensic Drug Measurements
Forensic science is the application of science to personal injury, murder, and other legal proceedings. Drug measurements in tissues obtained at autopsy or in other bodily fluids such as saliva, urine, and blood maybe useful if a suspect or victim has taken an overdose of a legal medication, has been poisoned, or has been using drugs of abuse such as opiates (eg, heroin), cocaine, or marijuana. The appearance of social drugs in blood, urine, and saliva drug analysis shows short term drug abuse. These drugs may be eliminated rapidly, making it more difficult to prove that the subject has been using drugs of abuse. The analysis for drugs of abuse in hair samples by a very sensitive assay methods, such as gas chromatography coupled with mass spectrometry provides information regarding past drug exposure. A study by Cone et al (1993) showed that the hair samples from subjects who were known drug abusers contained cocaine and 6-acetyl morphine, a metabolite of heroin (diacetylmorphine).
Significance of Measuring Plasma Drug Concentrations
The intensity of the pharmacologic or toxic effect of a drug is often related to the concentration of the drug at the receptor site, usually located in the tissue cells. Because most of the tissue cells are richly perfused with tissue fluids or plasma, measuring the plasma drug level is a responsive method of monitoring the course of therapy.
Clinically, individual variations in the pharmacokinetics of drugs are quite common. Monitoring the concentration of drugs in the blood or plasma ascertains that the calculated dose actually delivers the plasma level required for therapeutic effect. With some drugs, receptor expression and/or sensitivity in individuals varies, so monitoring of plasma levels is needed to distinguish the patient who is receiving too much of a drug from the patient who is supersensitive to the drug. Moreover, the patient’s physiologic functions may be affected by disease, nutrition, environment, concurrent drug therapy, and other factors. Pharmacokinetic models allow more accurate interpretation of the relationship between plasma drug levels and pharmacologic response.
In the absence of pharmacokinetic information, plasma drug levels are relatively useless for dosage adjustment. For example, suppose a single blood sample from a patient was assayed and found to contain 10 mg/mL. According to the literature, the maximum safe concentration of this drug is 15 mg/mL. In order to apply this information properly, it is important to know when the blood sample was drawn, what dose of the drug was given, and the route of administration. If the proper information is available, the use of pharmacokinetic equations and models may describe the blood level–time curve accurately and be used to modify dosing for that specific patient.
Monitoring of plasma drug concentrations allows for the adjustment of the drug dosage in order to individualize and optimize therapeutic drug regimens. When alterations in physiologic functions occur, monitoring plasma drug concentrations may provide a guide to the progress of the disease state and enable the investigator to modify the drug dosage accordingly. Clinically, sound medical judgmentand observation are most important. Therapeutic decisions should not be based solely on plasma drug concentrations.
In many cases, the pharmacodynamic response to the drug may be more important to measure than just the plasma drug concentration. For example, the electrophysiology of the heart, including an electrocardiogram (ECG), is important to assess in patients medicated with cardiotonic drugs such as digoxin. For an anticoagulant drug, such as dicumarol, prothrombin clotting time may indicate whether proper dosage was achieved. Most diabetic patients taking insulin will monitor their own blood or urine glucose levels. For drugs that act irreversibly at the receptor site, plasma drug concentrations may not accurately predict pharmacodynamic response. Drugs used in cancer chemotherapy often interfere with nucleic acid or protein biosynthesis to destroy tumor cells. For these drugs, the plasma drug concentration does not relate directly to the pharmacodynamic response. In this case, other pathophysiologic parameters and side effects are monitored in the patient to prevent adverse toxicity.