Pathogenesis of Cell Injury: Damage to cells can be either reversible or irreversible. The cellular reaction may be adaptive, and homeostasis is maintained, depending on the severity of the injury. When the degree of the injury (stress) exceeds the cell’s ability to repair itself, cell death occurs. The time of exposure to a damaging stimulus and the intensity of the damage caused are both factors that influence cell death. Internally controlled cell death, chromatin condensation, and fragmentation, or severe cell enlargement or cell rupture, denaturation and coagulation of cytoplasmic proteins, and destruction of cell organelles (necrosis) (apoptosis). Now that we’ve covered the causes of cell damage and necrosis, as well as their morphologic and functional correlates, we’ll look at the molecular foundation of cell injury in greater depth, before illustrating the key concepts with a few examples of common injuries. The biochemical mechanisms that link any given injury to the cellular and tissue manifestations that emerge are complicated, interrelated, and intricately braided with multiple intracellular metabolic pathways. As a result, pinpointing individual molecular modifications induced by a given assault might be challenging.
The cellular response to injurious stimuli depends on the type of injury, its duration, and its severity. Thus, low doses of toxins or a brief duration of ischemia may lead to reversible cell injury, whereas larger toxin doses or longer ischemic intervals may result in irreversible injury and cell death. The consequences of an injurious stimulus depend on the type, status, adaptability, and genetic makeup of the injured cell. The same injury has vastly different outcomes depending on the cell type; thus, striated skeletal muscle in the leg accommodates complete ischemia for 2 to 3 hours without irreversible injury, whereas cardiac muscle dies after only 20 to 30 minutes. The nutritional (or hormonal) status can also be important; clearly, a glycogen-replete hepatocyte will tolerate ischemia much better than one that has just burned its last glucose molecule. Genetically determined diversity in metabolic pathways can also be important. For instance, when exposed to the same dose of a toxin, individuals who inherit variants in genes encoding cytochrome P-450 may catabolize the toxin at different rates, leading to different outcomes. Much effort is now directed toward understanding the role of genetic polymorphisms in responses to drugs and toxins and in disease susceptibility.
Cell injury results from functional and biochemical abnormalities in one or more of several essential cellular components. The most important targets of injurious stimuli are
- Mitochondria, the sites of ATP generation;
- Cell membranes, on which the ionic and osmotic homeostasis of the cell and its organelles depends;
- Protein synthesis;
- The cytoskeleton;
- The genetic apparatus of the cell.

Table of Contents
A) Mechanism of Reversible Cell Injury
As the name implies, this happens when a cell is subjected to prolonged stress and is unable to adapt in order to cope. Cell injury that is reversible causes cellular and morphological alterations that can be reversed if the stimulus is removed. Reversible cell damage can be caused by one of three methods.
- Depleted resources of ATP in the cell owing to decreased levels of Oxidative Phosphorylation.
- Hydropic cellular swelling, a phenomenon caused by changes in ion concentrations and water influx.
- Organelles within the cell show minute alterations.
a) ATP depletion:
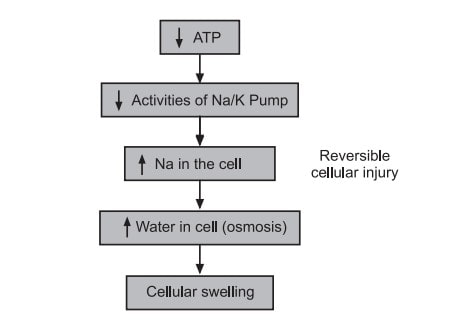
ATP depletion and decreased ATP synthesis are frequently associated with both hypoxic and chemical (toxic) injury. High-energy phosphate in the form of ATP is required for many synthetic and degradative processes within the cell. These include membrane transport, protein synthesis, lipogenesis, and the deacylation reacylation reactions necessary for phospholipid turnover. Depletion of ATP to 5% to 10% of normal levels has widespread effects on many critical cellular systems. The activity of the plasma membrane energy-dependent sodium pump is reduced. Failure of this active transport system, due to diminished ATP concentration and enhanced ATPase activity, causes sodium to accumulate intracellularly and potassium to diffuse out of the cell. The net gain of solute is accompanied by isosmotic gain of water, causing cell swelling, and dilation of the endoplasmic reticulum, and cellular energy metabolism is altered. If the supply of oxygen to cells is reduced, as in ischemia, oxidative phosphorylation ceases and cells rely on glycolysis for energy production. This switch to anaerobic metabolism is controlled by energy pathway metabolites acting on glycolytic enzymes.
Phosphofructokinase and phosphorylase activities are stimulated by a decrease in cellular ATP and a rise in adenosine monophosphate. As a result, the rate of anaerobic glycolysis increases, which is designed to keep the cell’s energy sources stable by creating ATP through the metabolism of glucose produced from glycogen. As a result, glycogen reserves are quickly depleted. The hydrolysis of phosphate esters leads in the buildup of lactic acid and inorganic phosphates during glycolysis. This lowers the intracellular pH, which causes numerous cellular enzymes to become less active.
b) Damage to Mitochondria:
Mitochondria provide the cell with life-sustaining energy in the form of ATP, but they also play a key role in cell damage and death. Mitochondria can be injured by an increase in cytosolic Ca2+, reactive oxygen species, and oxygen deprivation, making them vulnerable to a wide range of harmful stimuli, such as hypoxia and toxins.
There are two major consequences of mitochondrial damage:
• Mitochondrial damage often results in the formation of a high-conductance channel in the mitochondrial membrane, called the mitochondrial permeability transition pores. The opening of this channel leads to the loss of mitochondrial membrane potential and pH changes, resulting in failure of oxidative phosphorylation and progressive depletion of ATP, culminating in necrosis of the cell.
• The mitochondria also contain several proteins that are capable of activating apoptotic pathways, including cytochrome C (the major protein involved in electron transport). Increased permeability of the mitochondrial membrane may result in leakage of these proteins into the cytosol and death by apoptosis. Thus, cytochrome C plays a key dual role in cell survival and death. In its normal location inside mitochondria, it is essential for energy generation and the life of the cell, but when mitochondria are damaged so severely that cytochrome C leaks out, it signals cells to die.
c) Influx of Calcium
Ca2+ influx occurs when the Ca2+ pump fails, causing damage to a variety of cellular components. The structural disruption of the protein synthesising machinery occurs with prolonged or severe ATP depletion, characterised as ribosome detachment from the rough endoplasmic reticulum and dissociation of polysomes into monosomes, resulting in a reduction in protein synthesis. In the end, the mitochondrial and lysosomal membranes are irreversibly damaged, and the cell dies. Proteins can become misfolded in cells that are deprived of oxygen or glucose, and misfolded proteins activate a biological reaction known as the unfolded protein response, which can cause cell harm and even death. Protein misfolding can also occur when cells are exposed to stress, such as heat, or when enzymes and free radicals attack proteins.
1. Defects in membrane permeability: Early loss of selective membrane permeability leading ultimately to overt membrane damage is a consistent feature of most forms of cell injury. The plasma membrane can be damaged by ischemia, various microbial toxins, lytic complement components, and a variety of physical and chemical agents. Several biochemical mechanisms may contribute to membrane damage.
2. Decreased phospholipid synthesis: The production of phospholipids in cells may be reduced whenever there is a fall in ATP levels, leading to decreased energy dependent enzymatic activities. The reduced phospholipid synthesis may affect all cellular membranes including the mitochondria themselves, thus exacerbating the loss of ATP.
3. Increased phospholipid break-down: Severe cell injury is associated with increased degradation of membrane phospholipids, probably due to activation of endogenous phospholipases by increased levels of cytosolic Ca2+ .
4. ROS: Oxygen-free radicals cause injury to cell membranes by lipid peroxidation, discussed earlier.
5. Cytoskeletal abnormalities: Cytoskeletal filaments serve as anchors connecting the plasma membrane to the cell interior. Activation of proteases by increased cytosolic Ca2+ may cause damage to elements of the cytoskeleton.
6. Lipid breakdown products: These include unesterified free fatty acids, acyl carnitine, and lysophospholipids, catabolic products that are known to accumulate in injured cells as a result of phospholipid degradation. They have a detergent effect on membranes. They also either insert into the lipid bilayer of the membrane or exchange with membrane phospholipids, potentially causing changes in permeability and electrophysiologic alterations. The most important sites of membrane damage during cell injury are the mitochondrial membrane, the plasma membrane and membranes of lysosomes.
7. Mitochondrial membrane damage: As discussed above, damage to mitochondrial membranes results in decreased production of ATP, culminating in necrosis, and release of proteins that trigger apoptotic death.
8. Plasma membrane damage: Plasma membrane damage leads to loss of osmotic balance and influx of fluids and ions, as well as loss of cellular contents. The cells may also leak metabolites that are vital for the reconstitution of ATP, thus further depleting energy stores.
9. Injury to lysosomal membranes: Injury to lysosomal membranes results in leakage of their enzymes into the cytoplasm and activation of the acid hydrolases in the acidic intracellular pH of the injured (e.g., ischemic) cell. Lysosomes contain RNAases, DNAases, Proteases, Glucosidases, and other enzymes. Activation of these enzymes leads to enzymatic digestion of cell components, and the cells die by necrosis.
10. Damage to DNA and proteins: Cells have mechanisms that repair damage to DNA, but if this damage is too severe to be corrected (e.g., after radiation injury or oxidative stress), the cell initiates its suicide program and dies by apoptosis. A similar reaction is triggered by improperly folded proteins, which may be the result of inherited mutations or external triggers such as free radicals. Since these mechanisms of cell injury typically cause apoptosis.
11. Reduced protein synthesis: As a result of continued hypoxia, membranes of endoplasmic reticulum and Golgi apparatus swell up. Ribosomes are detached from granular endoplasmic reticulum and polysomes are degraded to monosomes, thus dispersing ribosomes in the cytoplasm and inactivating their function. Similarly reduced protein synthesis occurs in Golgi apparatus. Up to this point, withdrawal of acute stress that resulted in reversible cell injury can restore the cell to normal state.
B) Mechanism of Irreversible Cell Injury
Long-term reductions in oxygenated blood flow cause irreversible cellular structural and function damage. The reversible cell injury cycle continues, eventually leading to irreversible cell damage (cell death). The inability of a cell to correct mitochondrial and plasma membrane dysfunction after reperfusion or reoxygenation in irreversible cell injury. Aside from that, there is a continuing depletion of ATP in proteins, a drop in intracellular pH, and lysosomal enzyme leakage into the cytoplasm. These metabolic alterations affect the cell’s usual functioning, which are as follows:
1. Mitochondrial damage:
As a result of continued decrease in oxygenated blood supply, irreversible cell damage occurs and on reperfusion with injured cell, excess intracellular calcium collects in the mitochondria disabling its function. Morphologically, mitochondrial changes are vacuoles in the mitochondria and deposition of amorphous calcium salts in the mitochondrial matrix.
2. Membrane damage
Damage to plasma membrane loses its normal function is the most important event in irreversible cell injury. Due to damage of the plasma membrane, cytosolic influx of calcium in the cell increases. Calcium activates endogenous phospholipase. Activated phospholipase degrade membrane phospholipids progressively which are the main constituents of lipid bilayer membrane. Besides these, activation of lytic enzymes ATPase which causes further depletion of ATP leads to decrease in the synthesis of new phospholipid for replacement.
3. Cytoskeletal damage
Activated intracellular protease or by physical effect of cell swelling, damages of cytoskeleton may lead to irreversible cell membrane injury.
4. Nuclear damage:
The nucleoproteins are damaged by the activated lysosomal enzymes such as proteases and endonucleases. Irreversible damage to the nucleus can be in three forms.
- Pyknosis: Condensation and clumping of nucleus which becomes dark basophilic.
- Karyorrhexis: Nuclear fragmentation in to small bits dispersed in the cytoplasm.
- Karolysis: Dissolution of the nucleus.
5. Lysosomal damage, cell death and phagocytosis:
The lysosomal membranes are damaged and result in escape of lysosomal hydrolytic enzymes. These enzymes are activated due to lack of oxygen in the cell and acidic pH. These hydrolytic enzymes include, Hydrolase, protease, glycosidase phosphatase, lipase, amylase, RNAase and DNAase which on activation bring about enzymatic digestion of cellular components and hence cell death. The dead cell is eventually replaced by masses of phospholipids called myelin figures which are either phagocytosed by macrophages or there may be formation of calcium soaps. Liberated enzymes just mentioned leak across the abnormally permeable cell membrane into the serum, the estimation of which may be used as clinical parameters of cell death. In myocardial infarction, estimation of SGPT, LDH, CKMB and cardiac troponins useful guides for death of heart muscle.
Also Read: Etiology of Cell Injury