Table of Contents
Drug distribution
Drug distribution: Once a drug has gained access to the bloodstream, it gets distributed to other tissues that initially had no drug, concentration gradient being in the direction of plasma to tissues.
The extent and pattern of a drug’s distribution are determined by its:
- lipid solubility
- ionization at physiological pH (a function of its pKa)
- extent of binding to plasma and tissue proteins
- presence of tissue-specific transporters
- differences in regional blood flow.
Drug movement continues until an equilibrium is reached between unbound drugs in the plasma and unbound drugs in the tissue fluids. As a result of the removal, there is a simultaneous decrease in both.
Presuming that the body acts as a single homogeneous compartment with volume V into which the medication is instantly and uniformly dispersed, the apparent volume of distribution (V) is calculated.

In this example, 1000 mg of drug injected i.v. produces steady-state plasma concentration of 50 mg/L, apparent the volume of distribution is 20 L
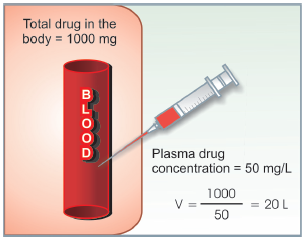
of distribution(V), Drug distribution
In the above example, the drug does not actually distribute into 20 L of body water, with the exclusion of the rest of it. This is only an apparent volume of distribution which can be defined as “the volume that would accommodate all the drug in the body if the concentration throughout was the same as in plasma”. The amount of drug present in the body as a multiple of that contained in a unit volume of plasma is a very useful pharmacokinetic concept.
Lipid-insoluble drugs do not enter cells V when they reach extracellular fluid volume, e.g. streptomycin, and gentamicin 0.25 L/kg
Drugs bound to plasma proteins are largely restricted to the vascular compartment and have low values, e.g. diclofenac and warfarin (99% bound) V = 0.15 L/kg. Drug distribution is not only a matter of dilution but also binding and sequestration.
A high V value suggests that there is a greater amount of drug in extravascular tissue. Because most of the drug is contained in other tissues and plasma concentration is low, drugs sequestered in other tissues may have V considerably higher than total body water or even body mass, e.g. digoxin 6 L/kg, propranolol 4 L/kg, morphine 3.5 L/kg. As a result, in the event of poisoning, medications with large distribution volumes are difficult to remove by hemodialysis.
Redistribution
Highly lipid-soluble medicines are first delivered to organs with high blood flow, such as the brain, heart, and kidney. Later, the medication is taken up by less vascular but bulkier tissues (muscle, fat)—plasma concentration declines, and the drug is removed from the highly perfused locations. If the drug’s site of action was in one of the highly perfused organs, redistribution causes the drug’s activity to stop. The greater the drug’s lipid solubility, the faster it gets redistributed.
Anaesthetic action of thiopentone sod. injected i.v. is terminated in few minutes due to redistribution.
Penetration into brain and CSF
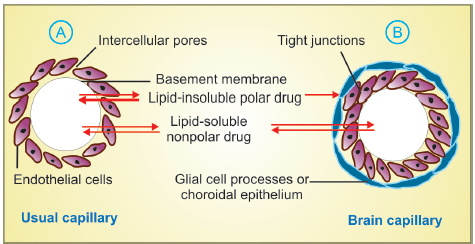
- Usual capillary with large paracellular spaces through which even large lipid-insoluble molecules diffuse.
- Capillary constituting blood brain or blood-CSF barrier. Tight junctions between capillary endothelial cells and investment of glial processes or choroidal epithelium do not allow passage of non lipid-soluble molecules/ions
The capillary endothelial cells in brain have tight junctions and lack large paracellular spaces. Further, an investment of neural tissue covers the capillaries. Together they constitute the so called blood-brain barrier (BBB).The choroid plexus has a comparable blood-CSF barrier: capillaries are bordered by choroidal epithelium with tight connections. Both of these barriers are lipoidal, preventing nonlipidsoluble medicines like streptomycin and neostigmine from entering.
Only lipid-soluble medicines can consequently enter the central nervous system and have an effect. Furthermore, efflux transporters found in the brain and choroidal arteries, such as P-gp and anion transporter (OATP), expel many medicines that reach the brain through other routes, enhancing the protective barrier against potentially hazardous xenobiotics. Dopamine does not reach the brain, but its precursor levodopa does, therefore it is used to treat Parkinson’s disease. The permeability of these barriers is increased when the meninges or brain are inflamed. It has been suggested that certain medicines accumulate in the brain via using natural chemical transporters.
There is also an enzymatic BBB: Monoamine oxidase (MAO), cholinesterase and some other enzymes are present in the capillary walls or in the cells lining them. They do not allow catecholamines, 5-HT, acetylcholine, etc. to enter brain in the active form.
Passage across placenta
Lipoidal placental membranes allow lipophilic medicines to flow freely while limiting hydrophilic drugs. The placental efflux P-gp, as well as other transporters like BCRP and MRP3, help to keep foetal exposure to medicines provided by the mother to a minimum. The placenta is also a location for drug processing, which may reduce or change the foetal’s exposure to the medication. Nonlipid-soluble medicines, on the other hand, acquire access to the foetus when they are present in large concentrations or for lengthy periods of time in maternal circulation.
Some influx transporters also operate at the placenta. Thus, it is an incomplete barrier and almost any drug taken by the mother can affect the foetus or the newborn (drug taken just before delivery, e.g. morphine).
Plasma protein binding
Most drugs possess physicochemical affinity for plasma proteins and are reversibly bound to them. Acidic drugs generally bind to plasma albumin and basic drugs to 1 acid glycoprotein. Binding to albumin is quantitatively more important. The extent of binding depends on the individual compound; no generalization for a pharmacological or chemical class can be made (even small chemical changes can markedly alter protein binding). For example, the binding percentage of some benzodiazepines is:
Flurazepam 10% Alprazolam 70%
Lorazepam 90% Diazepam 99%
Increased drug concentrations can saturate the binding sites, resulting in decreased fractional binding when large quantities of the medication are administered. The typical therapeutic plasma aconcentration of a medication is given as a percentage binding. The following are the clinical consequences of plasma protein binding:
- Highly plasma protein bound drugs are largely restricted to the vascular compartment because protein bound drugs do not cross membranes (except through large paracellular spaces, such as in capillaries). They tend to have smaller volumes of distribution.
- The bound fraction is not available for action. However, it is in equilibrium with the free drug in plasma and dissociates when the concentration of the latter is reduced due to elimination. Plasma protein binding thus tantamounts to temporary storage of the drug.
- A high degree of protein binding generally makes the drug long-acting, because the bound fraction is not available for metabolism or excretion, unless it is actively extracted by the liver or by the kidney tubules.
- The generally expressed plasma concentrations of the drug refer to bound as well as free drug.
The degree of protein binding should be taken into account while relating these to concentrations of the drug that are active in vitro, e.g. MIC of an antimicrobial. - One drug can bind to many sites on the albumin molecule. Conversely, more than one drug can bind to the same site. This can give rise to displacement interactions among drugs bound to the same site (s).
Make sure check our other amazing article: Drug bioavailability
Reference book: Essentials of Medical Pharmacology Seventh Edition by KD TRIPATHI MD